| Petit guide de bioinfo |
| Home page | UNIX | Assemblage | Alignement de séquences |
Points abordés
Introduction à l'assemblage de séquences
Depuis que les techniques de séquençage ont été proposées au milieu des années 1970 (Maxam et Gilbert, Sanger), des améliorations ont permis d'obtenir des lectures pouvant aller jusqu'à 1000 nucléotides. La longueur de ces fragments reste cependant sans aucune mesure avec les segments d'ADN que l'on souhaite séquencer aujourd'hui (génome de virus, génome de bactéries, BAC, ...). En 1982, Sanger et al. ont proposé une méthode connue aujourd'hui sous le nom de séquençage shotgun : on génère de manière aléatoire un ensemble de fragments d'une séquence cible et l'on tâche de reconstruire cette séquence en assemblant ces courts fragments sur la base de chevauchements de leur séquence de manière tout à fait comparable à l'assemblage des pièces d'un puzzle. Les fragments étant générés de manière aléatoire, la somme totale des fragments doit représenter plusieurs fois l'équivalent de la séquence cible (5X,8X) si l'on souhaite pouvoir reconstruire une proportion convenable de cette dernière. Même dans le cas d'une redondance importante (le séquençage du génome humain par Celera a utilisé une méthode de shotgun hierarchique avec une redondance de 10X), il faut s'attendre à ce que certaines régions de la région cible ne soit pas couverte par la séquence reconstruite et le résultat d'un d'assemblage sera alors consitué d'un ensemble de séquences (contigs) non chevauchantes. Dans le cas d'un séquençage d'une banque d'EST, il existe également une redondance qui résulte elle de la redondance des ARNm dans les tissus considérés. On ne recherche plus ici à reconstruire une seule séquence mais un ensemble de séquences qui représentent de manière plus ou moins fidèle l'ensemble des gènes qui s'expriment dans ces tissus. La redondance naturelle ou artifielle permet également d'identifier des segments présentant des variations et donc de détecter d'éventuels SNP. Enfin, dans le cas d'une démarche de séquençage sans redondance a priori mais utilisant un robot séquenceur, la comparaison 2 à 2 des séquences qui est réalisée au cours du processus d'assemblage permet de détecter d'éventuelles contaminations entre les puits des plaques traitées par le robot. En conclusion, il nous semble que la plupart des étapes décrites dans ce document devraient être réalisées en routine lorsque l'on traite des séquences brutes issues d'un séquenceur.
Lecture d'un chromatogramme
Les séquenceurs actuels fournissent, pour chaque fragment séquençé, un recodage des chromatogrammes sous la forme d'une trace ainsi que la séquence des nucléotides telle qu'elle est déterminée par un algorithme de lecture de traces ("base calling") généralement intégré aux fonctionnalités du séquenceur. Ewing et al. ont développé un autre algorithme de lecture des bases à partir d'une trace (cet algorithme est implémenté dans le logiciel Phred). Un analyse comparée des résultats de leur algorithme et de celui proposé par le séquenceur ABI (décrite dans Ewing et al.) indique que le premier réalise la lecture avec un taux d'erreur inférieur au second. Une autre qualité du logiciel Phred est qu'il fournit de plus pour chaque base lue une mesure de la qualité de la lecture. Cette mesure reflète la probalilité d'erreur de la lecture qui est estimée d'après l'aspect de la trace au voisinage de cette base: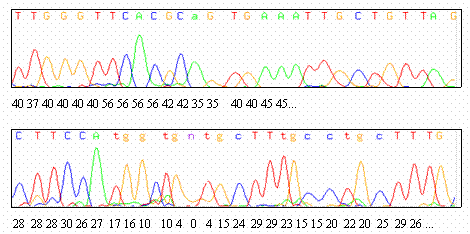

Le programme Phred
Principe
Le principe de l'algorithme de lecture du programme phred repose sur une technique courante du traitement du signal qui permet de détecter la périodicité du signal.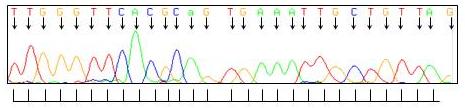
- Ewing B., Hillier L., Wendl M., and Green P. 1998. Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res: 8:175-185.
- Ewing B. and Green P. 1998. Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res: 8:186-194.
Fonctionnement
Le programme Phred lit des fichiers de trace issus de séquenceurs. Il peut lire les formats suivants (SCF, ABI et ESD). Il fournit en sortie la séquence des bases lues et une mesure de la qualité associée à chaque base. Phred peut traiter un seul fichier comme un ensemble de fichiers de trace. Il est également possible de choisr le format de sortie (pour plus de détail on peut se reporter à la documentation complète du programme documentation complète).Exemple 1
Supposons que vous souhaitiez lire les chromatogrammes qui se trouvent dans le répertoire "chromat_dir" et obtenir en sortie deux fichiers au format FASTA, l'un contenant l'ensemble des séquencs et l'autre les mesures de qualité (Ce sont ces deux fichiers qui seront nécessaire pour le lancement du programme Cap3 par exemple). Vous devez alors lancer le programme de la manière suivante :
mon_prompt_unix> phred -id chromat_dir -sa seqs_fasta -qa seqs_fasta.qual |
Exemple 2
Supposons que vous souhaitiez lire les chromatogrammes qui se trouvent dans le répertoire "chromat_dir" et lancer par la suite le programme phrap pour assembler les contigs et le programme consed pour les examiner. Le programme consed ayant besoin de fichiers de type phd, il vous faudra alors réaliser la lecture des chromatogtrammes en deux temps :
mon_prompt_unix> phred -id chromat_dir -pd phd_dir |
mon_prompt_unix> phd2fasta -id phd_dir -os seqs_fasta -oq seqs_fasta.screen.qual |
Elaguage des séquences
Si l'on souhaite utiliser les séquences autrement qu'avec les programmes d'assemblage Phrap et Cap3 (pour les comparer avec des séquences d'une banque par exemple), il est raisonnable de ne conserver que la partie de bonne qualité des fragments. Le programme phred offre deux possibilités, les options trim et trim_alt.- trim l'algorithme associé à cette option parcourt les séquences à partir des deux extrémités à l'aide d'une fenêtre de 50 bases et décale cette fenêtre tant que le nombre de bases avec une qualité inférieure à un certain seuil (par exemple 20) est supérieur à 5. La début de la séquence de bonne qualité se situe précisément à la première fenêtre pour laquelle ce nombre est inférieur à 5.
- trim_alt le principe est ici de sélectionner la plus longue sous-séquence qui possède un taux d'erreur inférieur à une valeur seuil fixée par défaut à 5%. Il est possible de modifier cette valeur seuil à l'aide de l'option trim_cutoff (voir la liste des options ci-dessous).
Exemple 3
Supposons que vous souhaitiez extraire les régions de bonne qualité pour l'ensemble des chromatogrammes se trouvant dans le répertoire chromat_dir :
mon_prompt_unix> phred -id chromat_dir -trim_alt "ATGGC" -tim_cutoff 0.01 -sa seqs_fasta -qa seqs_fasta.qual |
Les arguments du programme
Nous précisons ici quelques arguments et options du programme phred. Pour une liste exhaustive voir la documentation complète du programme.
Input Options ------------- -id nom_repertoire Lit les fichiers de trace situé dans le repertoire nom_repertoire. -if nom_fichier Lit les fichiers de trace listés dans le fichier de nom nom_fichier. ... Output Options -------------- -st fasta Le format de sortie est FASTA pour les séquences (Default.) -s Ecrit les fichiers de séquences avec un nom obtenu en ajoutant ".seq" aux noms des fichiers en entrée. Les fichiers sont stockés dans le répertoire courant. -sd nom_repertoire Ecrit les fichiers de séquences avec un nom obtenu en ajoutant ".seq" aux noms des fichiers en entrée. Les fichiers sont stockés dans le répertoire nom_repertoire. -sa nom_fichier Ecrit toutes les séquences au format FASTA dans un unique fichier de nom nom_fichier. -qt fasta Le format de sortie est FASTA pour les fichiers de qualité (Default.) -q Ecrit les fichiers de qualité avec un nom obtenu en ajoutant ".qual" aux noms des fichiers en entrée. Les fichiers sont stockés dans le répertoire courant. -qd nom_repertoire Ecrit les fichiers de qualité avec un nom obtenu en ajoutant ".qual" aux noms des fichiers en entrée. Les fichiers sont stockés dans le répertoire nom_repertoire. -qa nom_fichier Ecrit toutes les mesures de qualité au format FASTA dans un unique fichier de nom nom_fichier. Processing Options ------------------ -trim_alt enzyme_sequence Permet de sélectionner une sous-séquence de bonne qualité (voir ci-dessus). L'option trim_cutoff permet -de préciser le taux d'erreur d'erreur toléré. Il est possible de plus de spécifier une courte séquence (site de reconnaissance de l'enzyme de restriction) afin de préciser le début de l'insert. Vous pouvez indiquer un enzyme nul en fournissant un chaîne nulle "". -trim_cutoff valeur précise le taux d'ereur toléré pour -trim_alt. La valeur par défaut est 0.05. -trim_fasta Trim sequences written to sequence and quality value FASTA files. Set trimming information in the FASTA headers to reflect the high quality of the sequence, and append the string 'trimmed' to the header. -trim_scf Trim sequence, quality values, and base locations written to SCF file. Append the string 'trimmed' to the comments. -trim_phd Trim sequence, quality values, and base locations written to PHD files. Also set the first and last high quality base locations specified in the 'TRIM' comment field to the numbers of the first and last bases of the trimmed sequence (the first base in the sequence is base number zero). Finally set the error probability cutoff value in the 'TRIM' comment field to -1.00 to indicate that the sequence is trimmed, and that the trim points may be unrelated to the error probability cutoff value. -trim_out Trim information in the FASTA, SCF, and PHD output files. This is equivalent to specifying '-trim_fasta', '-trim_scf', and '-trim_phd' on the command line. -trim enzyme_sequence Perform sequence trimming on the current sequence. Bases are trimmed from the start and end of the sequence on the basis of trace quality. In addition, enzyme_sequence specifies a short base sequence (typically the recognition sequence of the restriction enzyme sequence used for subcloning) that is used to trim bases off the start of the current sequence. You can specify a NULL enzyme sequence using empty double quotes, "". We recommend against using this option because we consider it to be too conservative. See the note below on the effect of using the trim option. Divers ------ -doc Permet d'obtenir la documentation complète. -h, -help Permet d'obtenir les options de la ligne de commande. |
Masquage du vecteur
La démarche de séquençage implique souvent le clonage des fragments d'ADN au sein d'un vecteur. Les amorces étant alors définies dans le vecteur en amont de l'insert, il est fréquent, voire systématique, que les lectures contiennent des portions du vecteur. Il convient de masquer ces régions avant d'envisager l'exploitation de ces séquences à l'aide de programmes d'alignement ou d'assemblage. On peut également noter que la détection du marqueur et de l'amorce en amont de l'insert est un guage de qualité du produit de séquençage.Le programme cross_match
Principe
Le programme est simplement une implémentation d'un algorithme d'alignement (smith-watermann modifié, swat) associé à un outil qui masque les régions présentant une similitude significative avec les séquences du ou des vecteurs spécifiés. Il permet également de détecter des régions de faible complexité (une succession de A par exemple). Contrairement à ce que l'on aurait pu espérer, il n'utilise pas la mesure de qualité des lectures pour réaliser l'alignement de séquences.Usage
Le programme cross_match se lance de la manière suivante :
mon_prompt_unix> cross_match seq_file1 [seq_file2 ...] [-option valeur] |
Exemple 1
Supposons que l'on veuille masquer les séquences qui se trouvent dans un fichier de nom seqs_fasta sachant que la séquence du vecteur est contenue dans un fichier nommé vecteur.seq. On lancera alors la commande suivante:
mon_prompt_unix> cross_match seqs_fasta vecteur.seq -minmatch 12 -minscore 20 -screen > screen.out |
Remarque : Attention si vous envisagez d'utiliser un programme d'assemblage (Phrap ou Cap3) avec un fichier de séquences masquées par cross_match, il faudra renommer le fichier des mesures de qualité afin qu'il ait le même préfixe que ce lui des séquences:
mon_prompt_unix> cp seqs_fasta.qual seqs_fasta.screen.qual |
Analyse d'une sortie de crossmatch
Une sortie du programme cross_match commence par un récapitulatif des fichiers fournis en paramètres, les paramètres du programme d'alignement et des statistiques sur la qualité des séquences (ces statistiques sont fournis seulement s'il existe un fichier d'extension .qual pour le premier fichier de séquences,seqs_fasta.qual dans l'exemple précédent). Elle se poursuit par une description des régions de faible complexité :
Maximal single base matches (low complexity regions): Q9-2f_B07_9-2f_057.ab1 A Score: 23 Residues: 834 - 856 Q9-2f_E10_9-2f_071.ab1 A Score: 36 Residues: 704 - 755 Q9-2f_G11_9-2f_084.ab1 T Score: 38 Residues: 583 - 892 Q9-2f_G12_9-2f_088.ab1 A Score: 20 Residues: 779 - 819 |
42 0.00 2.17 0.00 Q11-1f_A03_11-1f_017.ab1 2 47 (711) vecteurg 94 140 (0) 43 0.00 0.00 2.08 Q11-1f_A06_11-1f_037.ab1 2 49 (722) vecteurg 94 140 (0) 47 0.00 0.00 0.00 Q11-1f_A07_11-1f_049.ab1 2 48 (730) vecteurg 94 140 (0) 47 0.00 0.00 0.00 Q11-1f_A08_11-1f_053.ab1 2 48 (727) vecteurg 94 140 (0) 47 0.00 0.00 0.00 Q11-1f_A09_11-1f_065.ab1 2 48 (732) vecteurg 94 140 (0) 42 0.00 2.17 0.00 Q11-1f_A11_11-1f_082.ab1 2 47 (730) vecteurg 94 140 (0) |
- score de l'alignement ;
- % de substitution sur le segment aligné ;
- % déletions dans la première séquence sur le segment aligné ;
- % insertions dans la première séquence sur le segment aligné ;
- nom de la séquence ;
- début du segment aligné sur la première séquence;
- fin du segment aligné sur la première séquence;
- nombre de bases au dela du segment aligné;
- Un C est précisé si la seconde séquence est complémentée;
- nom de la séquence du vecteur présentant une similitude;
- les trois dernières colonnes indiquent la position du segment dans la séquence du vecteur ;
Les arguments du programme
On utilise généralement le programme cross_match par l'intermédiaire du script phredPhrap (voir plus bas). Si toutefois vous souhaitez utiliser directement ce programe de masquage nous précisons quelquesuns de ses arguments et options. Pour une liste exhaustive voir la documentation complète du programme.On utilise
Input Options ------------- -minmatch n Précise qu'une similitude n'est considérée que si elle implique un minimum de n bases. -minscore s Précise qu'une similitude n'est considérée que si son score est d'au moins s. ... Output Options -------------- -screen si l'on soufaite produire un fichier de séquences avec les protions de vecteur masquées par des X. Le fichier des séquences masquées est nommé en rajoutant ".screen" au nom du fichier d'entrée. |
Assemblage des séquences
La grande majorité des programmes d'assemblage fonctionne sur le principe de trois étapes successives suivantes :- Comparaison de toutes les séquences 2 à 2 ;
- Agencement des séquences au sein de clusters ;
- Alignement multiple des séquecens pour chaque cluster et création d'une séquence consensus : le contig ;
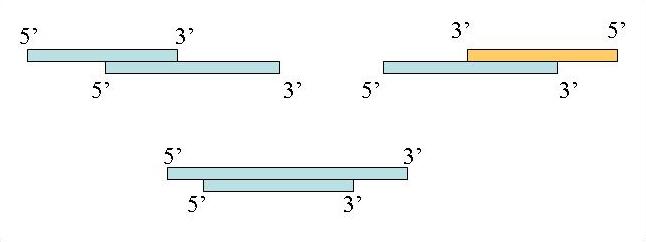
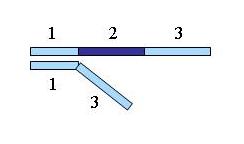
Une similitude de séquence comme celle décrite ci-dessous sera rejeté par le programme :
Le programme Phrap
Principe
Le programme phrap implémente la stratégie décrite ci-dessus. Il utilise une approche dîte gloutonne pour l'agencement des séquences. Le terme signifie que l'on procède par agglomération successive en commençant par assembler les lectures qui présentent la meilleure similarité. C'est l'approche que l'on utilise naturellement pour faire un puzzle, en commençant par les pièces dont l'assemblage ne fait pas de doute. Le programme phrap utilise les mesures de qualité des séquences pour évaluer la qualité d'un alignement :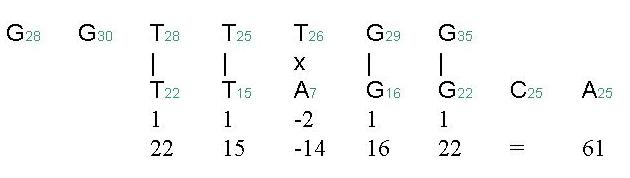
Usage
Le programme phrap se lance simplement de la manière suivante:
mon_prompt_unix> phrap seqs_fasta.screen -new_ace > phrap.out |
Analyse d'une sortie de phrap
Le programme phredPhrap
Le programme phredPhrap est un script perl qui lance successivement les différents programmes cités plus haut (phred, cross_match et phrap). Il permet donc à l'aide d'une seule commande d'assembler des lectures disponibles sous la forme de chromatogrammes. Afin de pouvoir utiliser le programme consed pour visualiser le résultat de l'assemblage il faut respecter une certaine arborescence de répertoires. Il faut créer une arborescence composée de 3 répertoire chromat_dir, edit_dir et phd_dir. Les chromatogrammes doivent se trouver dans le répertoire chromat_dir et le programme PhredPhrap se lance dans le répertoire edit_dir. On peut utiliser avec phredPhrap toutes les options du programme phrap ; elles seront transmises à ce programme lorsqu'il sera lancer ((pour plus de détail on peut se reporter à la documentation complète du programme documentation complète). PhredPhrap se lance donc de la manière suivante :
mon_prompt_unix/edit_dir> phredPhrap |
Les arguments du programme
Comme indiqué ci dessus ce programme peut se lancer sans arguments. Il est cependant possible d'utiliser toutes options du programme d'assemblage phrap qui lui seront transmis. Pour une liste exhaustive voir la documentation complète du programme.
Le programme Consed
Le programme consed permet, à l'aide d'une interface graphique, de visualiser les contigs produits par phrap. De manière plus général il permet de lire un fichier au format ace (extension .ace) qu'il soit crée par le programe phrap ou le programme cap3. Il faut dans les deux cas veiller à respecter l'arborescence des trois répertoires décrite plus haut.Documentation complète et officielle des programmes Phred, Phrap et Consed
On trouvera la documentation complète des programmes Phred, Phrap et Consed sur le site suivant:http://www.phrap.org
Le programme Cap3
Le programme PhredCap
Le programme PhredCap est tout à fait comparable au programme phredPhrap, il permet cependant quelques fonctionnalités supplémentaires. Vou pouvez trouver une documentation brève du programme à la page suivante : phredCap
Problèmes couramment rencontrés lors de l'assemblage
Voir les travaux pratiquesAgrégation abusive de séquences
Séparation abusive de contigs
© Groupe de Travail: Formation Interne BioInfo
Created 02/2003 .
Comments, suggestions, etc. Mail